
Special Report from ProPublica
by Marshall Allen and Olga Pierce, ProPublica
This story was co-published with the New York Times.
Carla Muss-Jacobs didn’t give much thought to the tools her surgeon would use to replace her knee. Like most patients, she just wanted to feel better and trusted that any devices in the operating room would be safe.

In her case, the surgeon sliced open her leg and positioned special cutting guides, like carpentry jigs, over her thigh and shin bones to line up his bone saw precisely. The device, called the OtisKnee, was supposed to speed the surgery and the recovery.
Muss-Jacobs’ recovery was not speedy. In terrible pain after the operation, she eventually underwent a second knee replacement.
As it turned out, the OtisMed Corporation, the maker of the OtisKnee, did not seek clearance from the Food and Drug Administration for its OtisKnee guides before it started selling them. When the company did apply for FDA review, its application was rejected because, the agency said, the company failed to show that the product was safe and effective.
In December, OtisMed and its former chief executive pleaded guilty in Federal District Court in Newark to criminal charges of distributing adulterated medical devices. The Justice Department said the company sold and distributed 18,000 of its OtisKnee devices from 2006 to 2009 without FDA approval.
No one can say with certainty if the OtisKnee device caused Muss-Jacobs’ problems, but in announcing an $80 million settlement of criminal and civil charges against OtisMed, United States Attorney Paul J. Fishman said patients:
“Should be entitled to trust that the devices their doctors are using are safe, effective, tested and approved.”
An examination of the OtisKnee case shows how easily that trust can be violated in the rapidly evolving world of medical devices, a thriving $110 billion-a-year industry. If not for a whistle-blower, the public might never have learned about the widespread use of a potentially dangerous device that sidestepped regulation.
About 700,000 knee replacements are performed every year, making it the most common elective surgery in the country. An aging population is increasing demand, creating opportunity for companies that make orthopedic devices and the accessories used to implant them.
OtisMed was an Alameda, Calif., start-up that saw an opening in that growing market. The company’s guides worked with knee replacement components made by other medical device makers. The idea was to use magnetic resonance imaging and three-dimensional software to create guides at an OtisMed facility that were then shipped to the hospitals. The guides directed the angle of the surgeon’s cuts so the artificial knee would be properly aligned. In theory, the method helped surgeons tailor bone cuts to a patient’s anatomy.
Experts say the cutting blocks were a good idea, and similar devices are made by other companies. But the FDA trusts manufacturers to properly classify their devices, effectively giving them say over whether safety studies are required before they are sold. That can allow some products that should receive closer scrutiny to slip by.
Dr. Steven B. Haas, chief of knee service at the Hospital for Special Surgery in Manhattan, said OtisMed pioneered the use of disposable, patient-specific surgical instruments. The problem was that research hadn’t demonstrated that the OtisMed alignment method was effective.
Dr. Haas said:
“OtisMed got it half right”
Feeling Like a Guinea Pig
In 2008, Muss-Jacobs, a real estate agent in Beaverton, Ore., had a terrible pain in her left knee. Years earlier, she had a knee operation, but things were getting worse, so she went to see Dr. Ronald Teed, an orthopedic surgeon.
Teed told her many doctors were using the OtisKnee device for custom knee replacements, with faster recoveries and reports of less pain, she said.
Teed performed the surgery in May 2008 at Tuality Community Hospital in nearby Hillsboro. Muss-Jacobs, now 56, said she awoke with so much pain she couldn’t stop crying. She knew immediately that something was wrong. Hospital records she provided document her persistent complaints about pain.
Muss-Jacobs said she had to hobble around with a walker and for several months was unable to show any homes to real estate clients. Six months later, she went to another surgeon, Dr. Ira Weintraub, in Portland, who said the new knee had failed and was misaligned. Dr. Weintraub performed a complex revision surgery, and Muss-Jacobs said she could walk the next day, though she still had a long, painful recovery. Teed said pain after a knee replacement could last up to a year and that Dr. Weintraub’s revision surgery wasn’t necessary. Dr. Weintraub and Tuality Community Hospital declined to comment.
Muss-Jacobs lived alone. Because she was unable to work, her bills mounted and, in 2009, she declared bankruptcy. When she couldn’t persuade a lawyer that a malpractice case was worthwhile, she sued Teed on her own, but the case was dismissed. (In an interview, Teed said Muss-Jacobs had refused to comply with her rehabilitation plan.
Teed said:
“She didn’t do what she was told and she ends up with a bad outcome.”
While Muss-Jacobs was preparing her lawsuit, she learned that there were complaints about the OtisKnee. She found news reports and OtisMed promotional materials and read medical device bulletin boards online. She discovered that some surgeons questioned whether the device was safe and that it had not been approved by the FDA.
The revelation made her feel like she’d been a “guinea pig,” she said.
Promoting the OtisKnee
Like other medical device makers, OtisMed marketed directly to providers. And it had a powerful message for them: The OtisKnee would simplify surgery and bring in extra revenue from additional MRI scans, according to Justice Department case filings.
The company made its pitch to doctors over dinner and during cocktail parties at meetings of the American Academy of Orthopedic Surgeons. The OtisKnee was supposed to preserve more of a patient’s bone and ligaments, improving fit and longevity. But none of the promotional claims had been evaluated by the FDA, the Justice Department said.
The company’s public relations efforts worked. According to a case study in PR Week, the number of surgeons adopting the OtisKnee doubled each month in 2007.
Among the converts was Teed.
He endorsed the OtisKnee to a gathering of potential patients in an informational video obtained by Muss-Jacobs. The surgery is a “piece of cake”
Teed tells the audience:
“It doesn’t take me a lot of effort.”
Teed explained that operating room efficiency is crucial to reducing the risk of infections and other complications. He said he had been able to do as many as 12 knee replacements a day by bouncing among two or three operating rooms. Then OtisMed approached him and said that with the OtisKnee, he could work even faster.
Teed says in the video:
“And of course, you can catch my attention with that, It’s like, Well, that sounds good to me, let’s sit down and talk.'”
Eventually, though, Teed said, he found problems. He said he had taken part in an OtisMed study and was one of the first surgeons to discover that the procedures were not always working as hoped.
He said:
“Some did. Some didn’t, They weren’t perfect.”
During some surgeries, he could see that alignments weren’t right. In a few cases he had to revert to the standard knee replacement method and redo the operation on the spot, he said.
He said he had visited the company’s headquarters and “they seemed like they couldn’t figure it out.” Later, Teed said, he heard that OtisMed had changed the materials used to make the cutting guides, which might have led to them warping when they were sterilized before surgery.
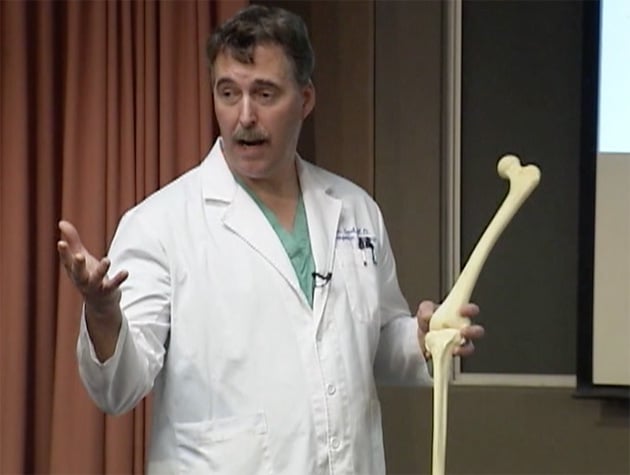
Surgeon Ronald Teed, shown in a screenshot taken from a video of an informational presentation, describes the advantages of the OtisKnee. (Tuality Healthcare)
The FDA should have been sterilizing “the crud” out of those things to test them, Teed said.
Faulty cutting guides were not a problem in Muss-Jacobs’ case, Teed maintained. Nonetheless, he said he had learned a valuable lesson from the OtisKnee:
“Don’t jump on the bandwagon too early.”
A Regulatory Bypass
The FDA regulates medical devices by classes, and each is treated differently.
Class III devices, like pacemakers, require extensive testing because they are implanted or sustain or support life and could put patients at serious risk.
Class II devices, like powered wheelchairs or pregnancy tests, are approved if companies assure that they are similar to other devices on the market.
Class I devices — bandages, dental floss, forceps and the like — must be registered but don’t require premarket review because they present a low risk. They can be sold without any other FDA involvement.
The FDA gives companies some freedom in classifying their devices. And in the case of the OtisKnee, OtisMed officials told doctors and hospitals that its cutting guides were a Class I device and didn’t need FDA approval or clearance, the Justice Department said.

Carla Muss-Jacobs has stacks of paperwork and bills from her knee surgeries.
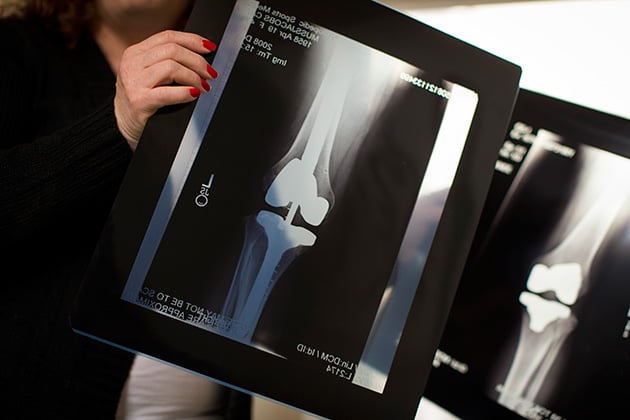
X-rays show Muss-Jacobs’ left knee after the joint was replaced using a device that the FDA hadn’t approved, right, and after a revision that required inserting metal rods into her femur (upper leg) and tibia (lower leg). (Leah Nash/Special to ProPublica)
According to the agency’s legal filings, OtisMed worked closely with Stryker, a $9 billion medical device company based in Kalamazoo, Mich., to market the OtisKnee. Stryker made knee replacement components that worked with the OtisKnee. Stryker became interested in buying OtisMed, according to the Justice Department case, and requested that the start-up apply for FDA clearance to market the OtisKnee, which it did in October 2008.
Nearly a year later, in September 2009, Stryker was prepared to acquire OtisMed for $100 million when the FDA notified OtisMed that it had not demonstrated that the guides were safe. The notice said that the OtisKnee was a Class III device and that OtisMed’s submission was missing data about how patients had fared, raising concerns about failure rates. The notice expressly warned against distributing the devices, according to the Justice Department.
The OtisMed board voted unanimously to halt shipments, the Justice Department said in court filings. But Charlie Chi, who was then the company’s chief executive, directed employees to ship a batch of guides that had been held up since the FDA denial. OtisMed sent out 218 of the devices to surgeons, the Justice Department filings state.
Stryker executives were unaware of the shipment, the filings say. Stryker’s acquisition of OtisMed went through in November 2009.
On Oct. 2, 2009, Richard Adrian, who worked in sales for Stryker, filed a whistle-blower case, accusing OtisMed of selling unapproved devices. Mr. Adrian received about $7 million as part of the resolution of the case. Mr. Adrian’s lawyer said he declined to comment.
In settling the case, OtisMed paid $80 million in criminal and civil fines. Mr. Chi is to be sentenced on March 18 and faces up to three years in prison and $300,000 in fines. Stryker was not charged with wrongdoing and agreed to audit its other devices to ensure they had proper FDA approvals.
Representatives from OtisMed, including Mr. Chi, declined to comment for this article. Stryker said in a statement that the criminal conduct occurred before it acquired OtisMed and without its previous knowledge, and that the company “is committed to conducting its affairs ethically and lawfully.”
Malfunction Reports
OtisMed sold $27 million worth of its cutting guides before the FDA rejected them. Problems began showing up as early as November 2007, according to a ProPublica review of injuries and side effects reported to the FDA.
There were 11 reports that month alone and 58 overall by the end of 2009, most classified as malfunctions:
“Femoral guide does not fit, rocks freely.” “Too much anterior slope in the tibia cut.” “Femur and tibia guides would not seat correctly. Both slid around freely.”
There were warning signs in the medical community, too. A January 2008 study in The Journal of Arthroplasty examined four cases: The study said.
“The potential for malalignment with this system places implants at high risk of early failure.”
No one knows how many patients might have been harmed by the device. Lawyers for the Justice Department said finding a number wasn’t part of the agency’s case.

Stacie Bilek, who directs premarket compliance at the FDA’s Center for Devices and Radiological Health, would not discuss the OtisKnee case because it had been the subject of a criminal investigation. But there are so many Class I medical devices on the market that the agency generally does not check to see if they are properly classified, she said:
“It’s based on voluntary compliance”
Ms. Bilek said, noting that the FDA will sometimes investigate complaints:
“We presume companies know the rules and regulations.”
An FDA spokesman said the agency learned in late 2007 that the OtisKnee was being sold without proper clearance. An investigation was opened, then dropped after OtisMed sought approval in 2008.
Diana Zuckerman, president of the National Center for Health Research, an FDA watchdog group, said the agency wasn’t doing enough to keep patients safe.
The FDA’s “surveillance system is so flawed and so weak and so subjective and so inadequate.
” Ms. Zuckerman said, “that there are no automatic red flags going up when something happens.”
As for Muss-Jacobs, December’s settlement was bittersweet. While she is happy that OtisMed paid a price, she was disappointed that no medical provider was taken to account in her case.
Muss-Jacobs asked:
“What happens to all the patients who had this device used on them? We’re so marginalized. It’s like we’re not even in the equation in all of this.”
Related stories: Read more patient safety coverage by Marshall Allen and Olga Pierce.
ProPublica is a Pulitzer Prize-winning investigative newsroom. Sign up for their newsletter.




